Chi sono?
Beh difficile dirlo, figuriamoci per voi immaginarselo….
Sono Nadia ho 18anni e mi sono da poco diplomata al liceo artistico.
Un ottimo risultato direi…mi hanno buttato fuori dalla scuola con un meritato 86.
Non mi posso lamentare di nulla non c’è che dire…ed anni ed anni di studio,
sono stati premiati.
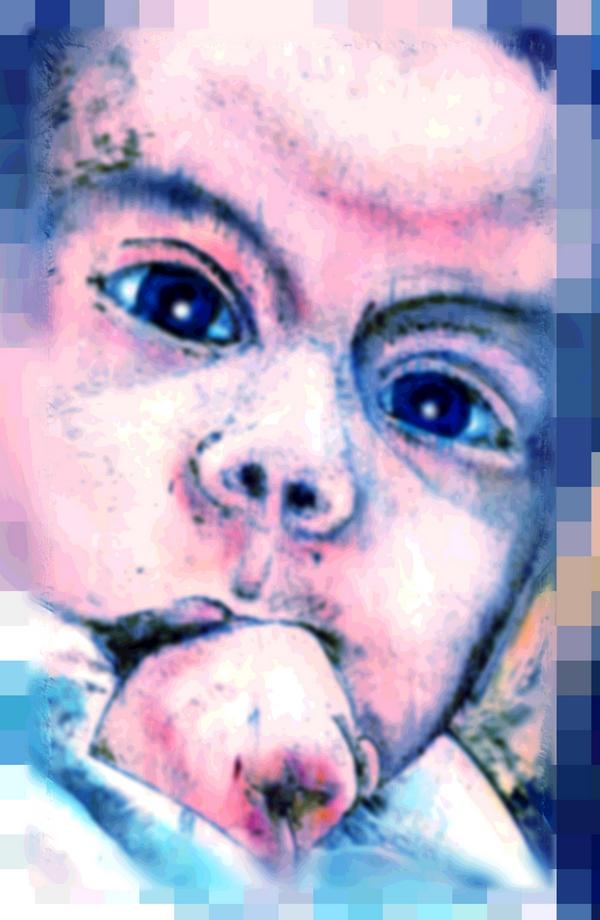
Qualche frutto della mia arte lo trovate anche qui nello space…anche la foto nello space è un mio lavoro…
Già la foto nello space…vi direte ma chi è?
Chiunque di voi a speso almeno 5 minuti in giro per lo space si sarà accorto di uno space dedicato ad un piccolo angioletto andato via...il piccolo Gabriele Perez, l’idea è stata del suo papà Luca Perez, un mio Grandissimo amico, che poi pian piano l’ho aiutato ha svilupparla…
Vi direte di nuovo…ma qual è questa idea??
Abbiamo dedicato un intero space dedicato al piccolo Gabry e alla sua storia…
Lui ci ha lasciati per colpa di una brutta malattia.
..La SMA..(Atrofia Muscolare Spinale), una malattia genetica che viene trasmessa dai genitori,
che sono portatori sani….insomma un po’ come la talassemia e che della SMA nessuno parla…
.. ..
il piccolo Gabry è andato via a soli 4 mesi dalla sua nascita il 10 Gennaio 2008, con la sua ultima crisi respiratoria…
e sì…perché questi bimbi affetti dalla SMA soffrono con le crisi respiratorie, non possono camminare e tanti medici pazzi pensano che per guarirli basti fare la fisioterapia…si la fisioterapia ad un bambino di 10 giorni(come ci ha raccontato una nostra amica mamma, che la stessa malattia a portato via il suo cucciolo a soli due mesi il piccolo Nathan).
.. ..
Chi ha un po’ di cuore si affeziona a tutte queste storie ed a tutti questi angioletti andati via e tutti gli altri che lottano per farcela…
.. ..
Io personalmente non ho avuto modo di conoscere il piccolo Gabriele ma solo a guardarlo nelle foto, si vede lo sguardo di un piccolo bimbo furbo, intelligente e vivace, che risponde pienamente agli stimoli della vita che si lascia trascinare dalle cose strane e curiose che lo circondano…
Era questo Gabriele Perez un piccolo cucciolo che giocava col suo papà e che lo rendeva felice, lui voleva farcela ma non gli è stato permesso…
Non gli è stato permesso a causa della scarsa informazione medica che ci qui a Palermo su questa malattia(come del resto anche nella parte restante dell’Italia).
La maggior parte dei medici ai bimbi che sono affetti da SMA1 li dà come “condannati a morte”, mentre una piccola parte di loro fa di tutto per salvarli, è così si hanno casi di SMA1 vivi in america ad 11anni….quindi non è proprio un’utopia far vivere questi piccoli angioletti.
.. ..
Tanti ne sono sopravvissuti e non sono stati dati come “già morti” dalle cartelle cliniche, eppure i medici che fanno questo operato vengono chiamati pirati…perché pirati? Perché quei santi di uomini sono riusciti nell’impossibile? Forse perché hanno fatto qualche ricerca in più? Forse perché sono stati i primi a non praticare la traectomia e sono riusciti a farli respirare con un metodo di ventilazione non invasiva?
Per questo sono dei pirati….
Si dovrebbero santificare uomini cosi!
Perché il governo non si occupa di finanziare la ricerca per questa malattia genetica?
Pian piano si sta espandendo a macchia d’olio e i portatori sani sono più di quanto si pensi, orami veramente si sta diffondendo come la talassemia, eppure qui in Italia sembra che tutti non se ne accorgano…
.. ..
Purtroppo se non agiscono quei poveri genitori che si sono ritrovati dall’oggi al domani portatori di SMA, senza che ne sappiano nulla, che si ritrovino “”assassini”” dei loro figli senza che loro potessero fare nulla; Qui in Italia non si smuoverebbe nulla…è la cruda verità.
.. ..
E poi…se tutta l’altra gente che dice di non c’entrarci nulla con tutto questo, sia più sensibile(perché bisogna ricordarsi che un giorno chiunque potrebbe ritrovarsi nella stessa situazione)le cose cambierebbero di più….questa malattia, come la talassemia, la sclerosi, la fibrosi cistica…verrebbe conosciuta alla pari di queste e qualcosa in più la si potrebbe ottenere.
.. ..
Io sono insieme a Luca Perez ed a Giuseppe Cavarerretta l’organizzatrici della serata di beneficenza “Gabriele Perez per gli affetti dell’Atrofia Muscolare Spinale(SMA)2 che si terrà a Paleria il & settembre 2008 alla Tonnara Florio.
.. ..
È un evento grande che coinvolgerà una miriade di persone importanti e non, coinvolgerà anche le tante famiglie che hanno subito il dolore di perdere un bimbo con ..la SMA.. e tutti quelli che hanno un bimbo vivo con ..la SMA..(perché ..la SMA.. nelle sue forme minori ossia ..la SMA..2 e ..la SMA..3 non è mortale…)racconteranno la loro esperienza, perche questi piccoli angioletti che sono volati in cielo hanno bisogno di una voce che li tenga vivi…e quella voce siamo tutti noi che ne teniamo il ricordo e lo rendiamo pubblico alla gente.
.. ..
Io da esterna rendo pubblica la storia del piccolo angioletto Gabriele…perchè la sua memoria resti viva nei nostri cuori, perché conosco il dolore del suo papà che un gran ragazzo dal cuore d'oro e che nella vita non ha mai avuto la giusta felicità che si merita...un pò amareggiato da essa e un pò insicuro e timoro di sè e di quello che lo circonda, una vita veramnete ingiusta nei suoi riguardi ed io che lo conosco lo posso giurare cento volte e so anche tutto il gran da farsi che si sta dando per aiutare tutti gli altri angioletti…ma presto vi presenterò tante altre storie di tanti altri piccoli angioletti in giro per il mondo allegandovi i loro piccoli spazi dove le loro mamme raccontano la loro storia.
.. ..
A presto Nadia…che supporta il piccolo angioletto Gabriele vhe ci guarda da lassù…
Non ti preoccupare! Qui le cose si stanno muovendo perché la tua ascesa in cielo non sia vana…
Ti voglio tanto bene piccolino…
e se la mia arte dovesse in qualche modo aiutare i nostri angioletti...io metto a disposizione la mia
abilità a servizio degli angeli.
.. ..
.. ..
.. ..